The post will only be a summary of some knowledge that the team gain during the past week or so about the phage and the genome of the phage that we were assigned.
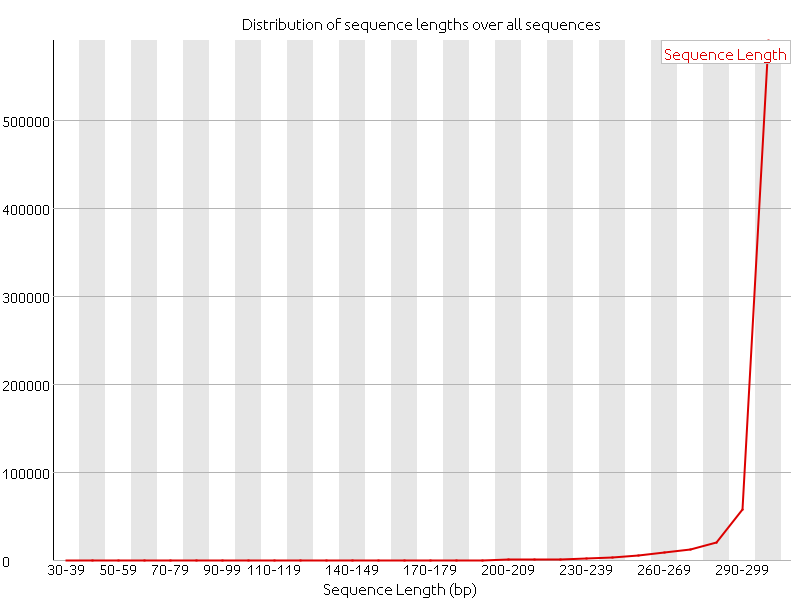
First of all, out of the ten assemblies we decided to continue with the one that was assembled with SPAdes, where all reads where given as input and the setting careful was used. This resulted in an assembly with 34 contigs, but where two of the contigs make up the majority of the total sizes. One of these contigs is 90000 bp and the other one is 76000 bp. The rest of the contigs are about 3000 bp or shorter. This makes us think that maybe one of the two larger contigs might be the phage genome. Later it was confirmed by Anders Nilsson that both of these contigs are phage genomes and from two different phages. The the smaller contig is the genome that the research group was looking for, and so in the continued work of this project we have started to characterize and annotate this genome. Anders also mentioned that this phage is virulent and so we expect and have to some extent confirmed that this phage has its own enzymes and mechanisms for replication. This was done by using BLAST against other phage genomes. We also were informed by Anders that this phage belongs to the family of P7 phages. Further annotation should, thus, be proceeded by using BLAST against genomes belonging this this family of phages, as far as possible.
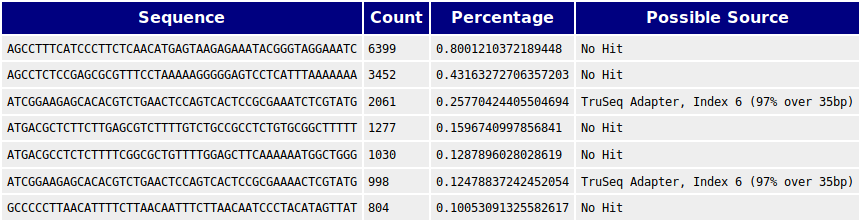
We also performed BLASTs against human and E. coli genomes and found matches against the genomes of these species that could not be found in human and E. coli. We thus concluded that there is contamination from these species in the samples. These are non the less irrelevant as we have been provided confirmation that the 76000 bp contig is the genome of the phage of interest. But this contig should be BLASTed against E. coli and human to assess if there might be reads from these species that might have been incorporated into the contig.
We decided to divided the work among us, where one of us would do research on the phage biology of our phage of interest and compare it to other phages as one means of characterization of our phage genome, one would research the ORFs of the phage genomes to try and predict unique genes for this phage, and one of us would do the research and testing necessary to find the terminal repeats of the genome. For my part I was given the task of finding the terminal repeats. This work will be conducted by researching the literature to find phages of the same family where the terminals have already been found to find clues about the terminal repeats, and also the coverage of the reads back to the genome should give clues about the position of the terminals, since it can be assumed that the repeats of the terminal have a higher coverage compared to the rest of the genome.
We have been looking for a tool to visualize the contigs and allow us to work with the genome in a visual manner, and Anders recommended the software Geneious. This software is a commercial software, but there is a possibility to get a 14 days free trial version. This is the software I will use to explore the terminal repeats.