Since we realized that we need to quality controls of the reads and remove adapters and trimming and filtering we have found the software FastQC which is a quality control tool for high throughput data. FastQC does not modify the reads. It just give different kinds of graphs that report the quality of the reads.
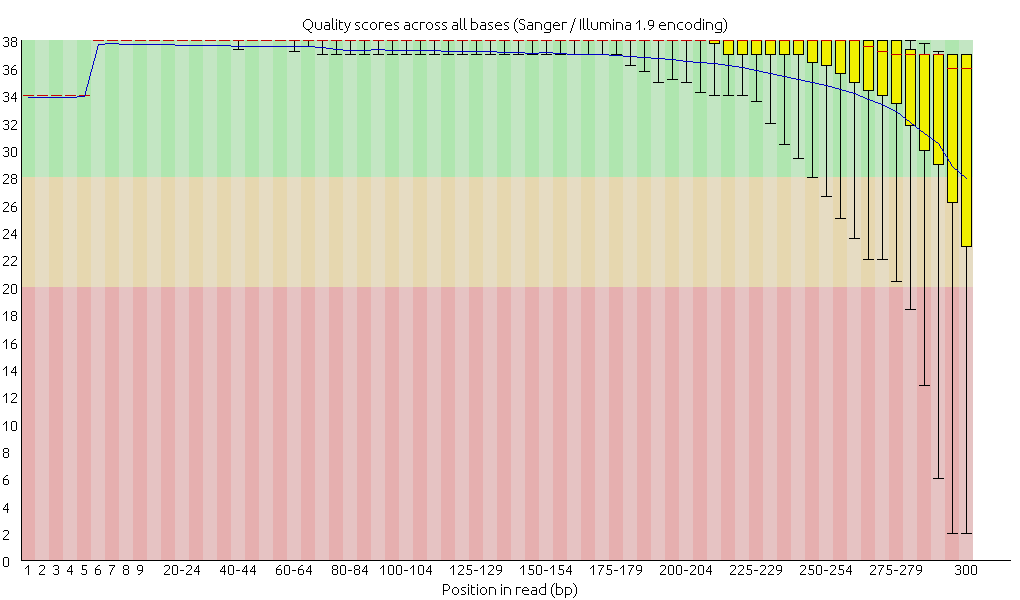
We analyzed the original dataset of reads with FastQC and the quality of the data is reported as good even though some categories give the failure and warnings. The per base sequence quality shows that the general quality of the bases is good even though it starts to drop by the end of the reads.
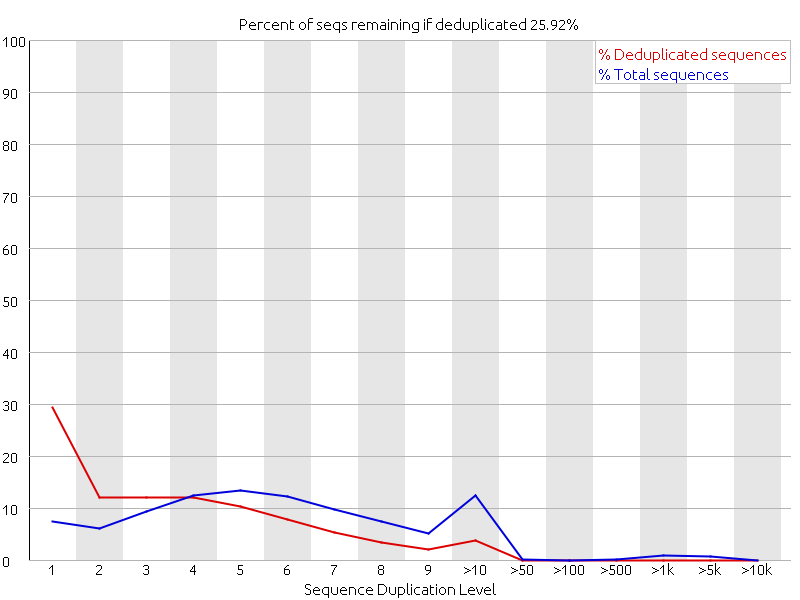
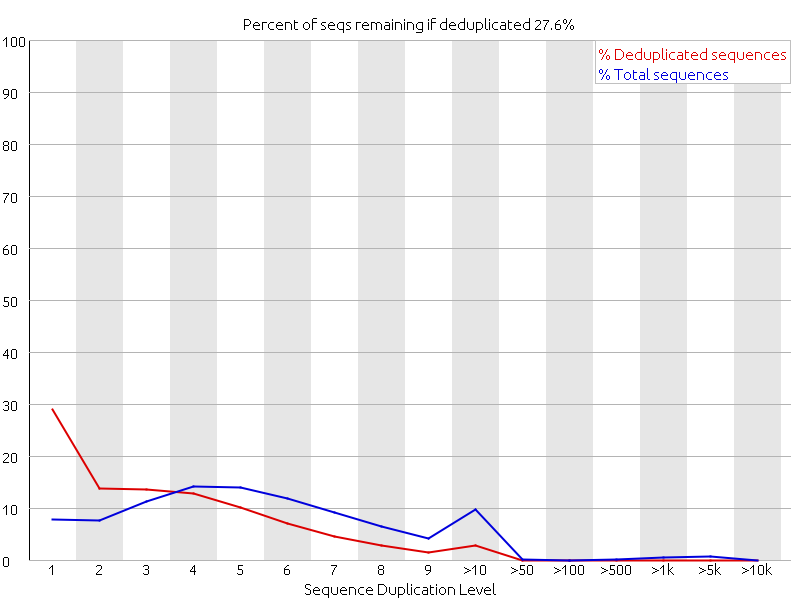
The category for sequence duplication levels is the only category that gives a failure. This may indicate some kind of enrichment.
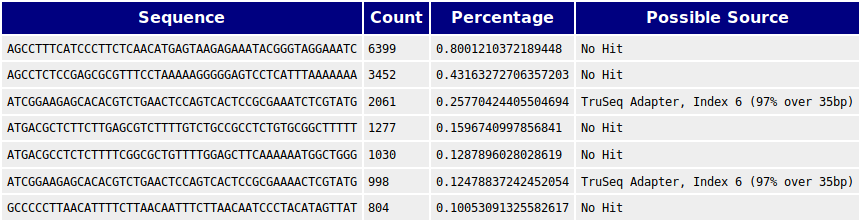
The category for over-represented sequences gives a warning. A sequence is regarded as over-represented and the software will raise a warning if the sequence makes up more than 0.1% of the total number of sequences. An over-represented sequence may be due to biological importance or to contamination. In the table it can be seen that the over-representation of two of the sequences are due to the Illumina adapter sequence.
The category for adapter content also gives a warning. The graph shows, though, that the source of the warning is a significant amount of Illumina adapter sequences.
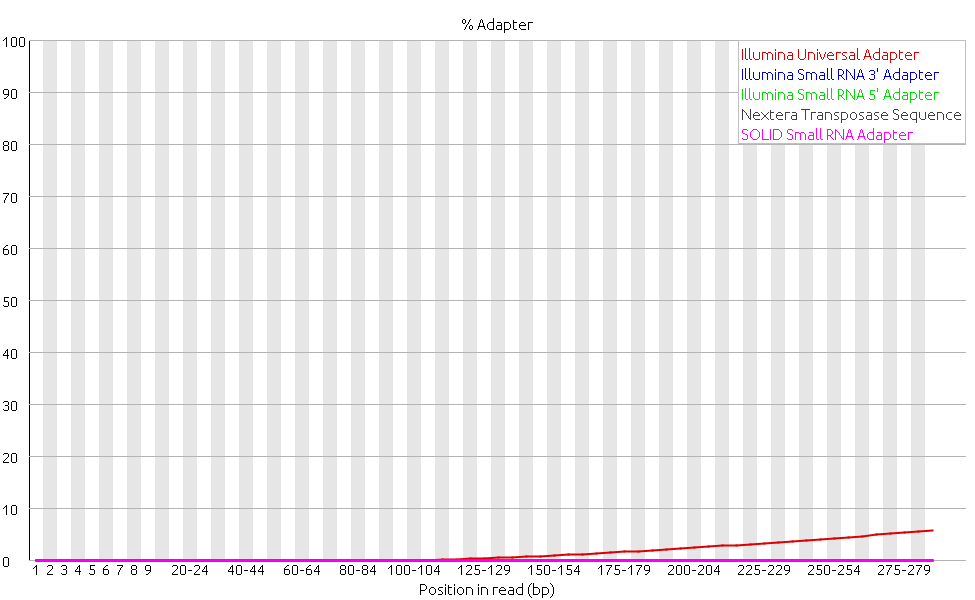
The analysis of the raw reads shows that there is a significant amount of Illumina adapter sequences in the dataset and thus adapter removal should be performed. This was previously done with Trimmomatic and the resulting reads were again analyzed with FastQC.
The per base quality improved drastically compared to before the removal of the adapters as seen in the image below.

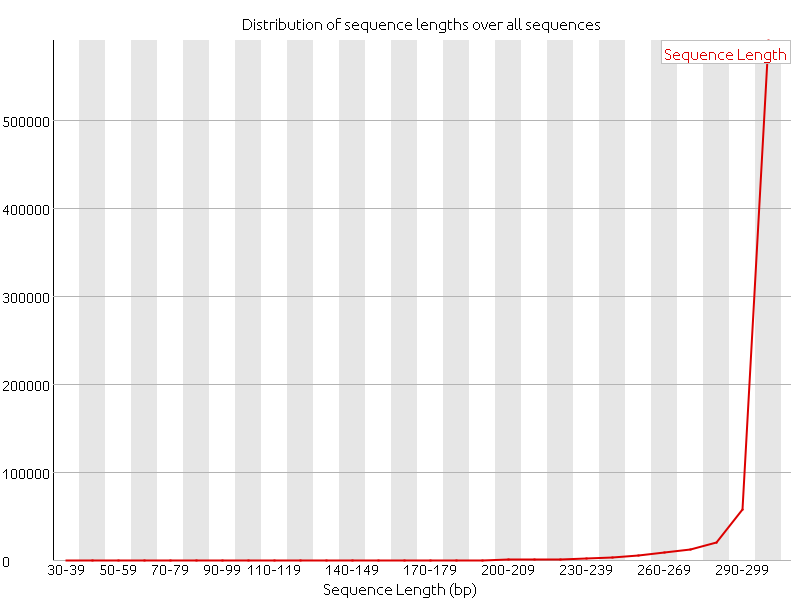
Before the adapter removal the distribution of sequence lengths had a perfect score since all the sequences where 300 nucleotides. After the adapters were removed the distribution of sequence lengths changed, as expected. Most of the sequences are very close to 300 nucleotides. According to the FastQC manual the software raises a warning as if all the sequences are not the same length. But this should not be a big issue in this case.
The sequence duplication levels did not change much after the removal of the adapters compared to before the adapter removal and still raises a failure, indicating an enrichment bias. The software raises a failure if more than 50% of the total amount of sequences are non-unique. I’m not sure if this will cause an issue with the assembly but we decided to continue with the next steps without looking closer into this issue.
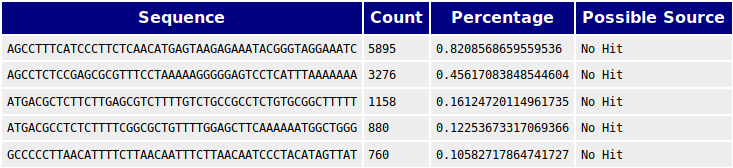
In the table of overrepresented sequences it can be seen that the sequences of the adapter have been removed, as expected. The rest of them from before the removal are still present and their sources are still unknown but should probably be BLASTed to find out more about their importance.
Finally, as expected the graph for adapter content shows that all the adapters were removed. This category turned from a warning to a pass.
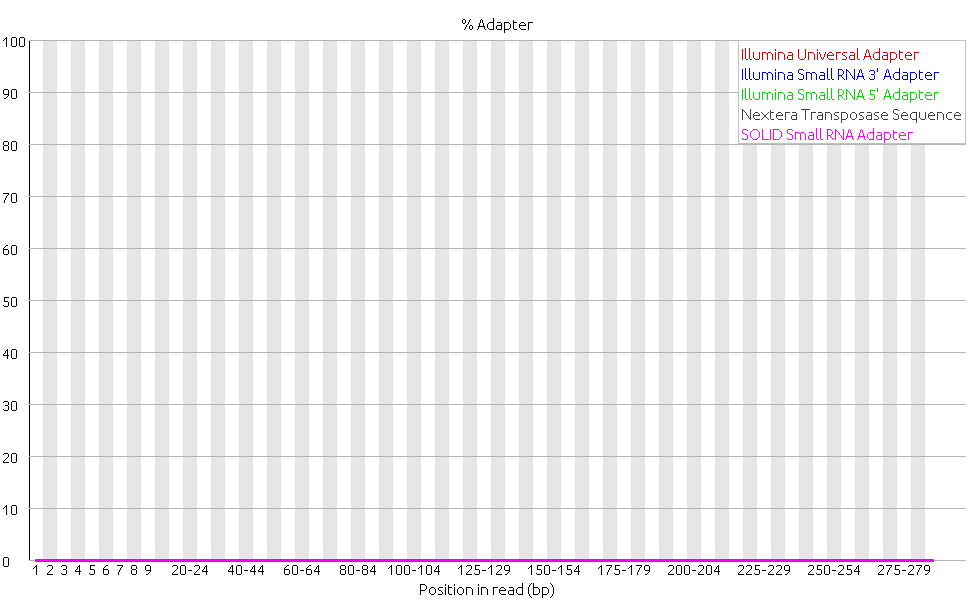
In conclusion we decided that the adapter removal improved the data quality and decided to continue with the adapter removed reads.