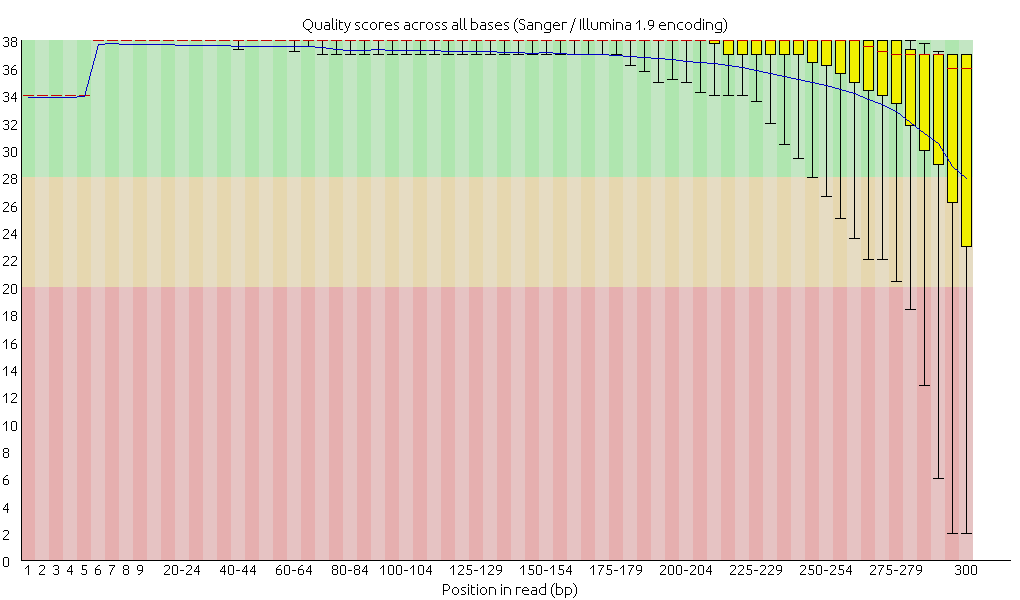
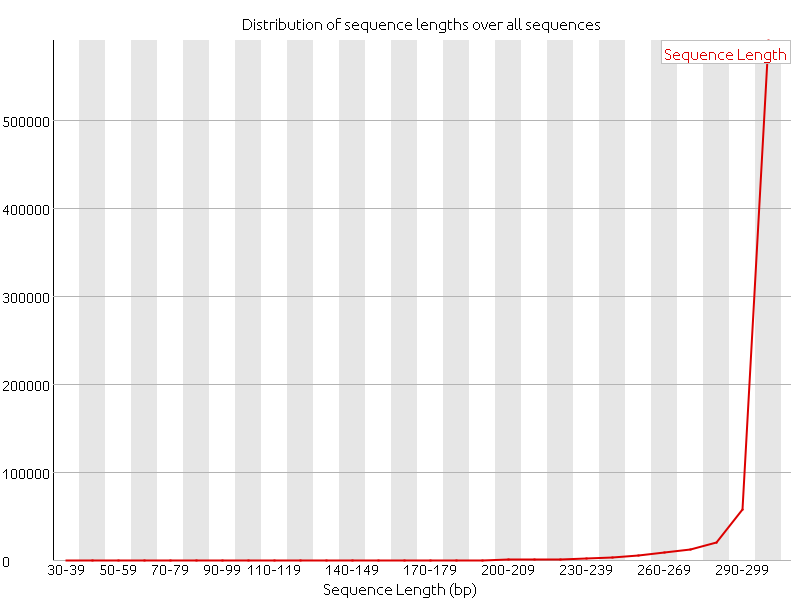
Last week we focused on doing more quality controls, and we started with doing alignments. After the adapter of the reads had been removed a quality control of the reads with FastQC clearly showed an increase in the quality of the data.
Then we proceeded to do trim and filter the reads with FastX. Trimming is when bases with poor quality are removed, and filtering is when entire reads of poor quality are removed from the dataset, either due to poor average quality, ambiguous base calling or short length. FastX is a collection of command line tools for pre-processing short reads. Using FastX to filter the data did not result in any changes of the quality of the data. The file sizes of the fastq files before and after remained the same and analyzing the files with FastQC confirmed this since there were no changes in the plots compared to the plots before filtering. The per base quality plot below is given as an example of no changes to before the filtering.

We decided to continue without the filtered reads.
Next we tried assembling the reads into contigs with two different assemblers: Velvet and SPAdes. After discussion with Anders Nilsson and consulting documentation from Illumina it was decided to assemble the reads with Velvet with three different k-mer sizes: 21, 41 and 61. SPAdes has an algorithm for calculating the k-mer size. In the literature it is also recommended to assemble with a lower coverage that is common when assembling phage genomes. This is because phage genomes are small and with a high coverage there is a risk that systematic errors would be treated as natural variations. For this reason the reads were also assembled with only 10% of the reads, for both Velvet and for SPAdes. In addition, when the reads were assembled with SPAdes they were assemble with the setting “careful” on and off. In total ten assemblies were made using SPAdes and Velvet.
The software Quast was used to analyze and require metrics for all assemblies. The result reported by Quast was (only contigs > 500 bp reported):
Velvet 21 k-mer:
Number of contigs: 32
Largest contig: 2926 bp
N50: 749 contigs
Total length: 24391 bp
Velvet 41 k-mer:
Number of contigs: 43
Largest contig: 1730 bp
N50: 711 contigs
Total length: 32159 bp
Velvet 61 k-mer:
Number of contigs: 63
Largest contig: 1241 bp
N50: 660 contigs
Total length: 42473 bp
Velvet 21 k-mer (10% coverage):
Number of contigs: 63
Largest contig: 5390 bp
N50: 1862 contigs
Total length: 85004 bp
Velvet 41 k-mer (10% coverage):
Number of contigs: 64
Largest contig: 7011 bp
N50: 2777 contigs
Total length: 100054 bp
Velvet 61 k-mer (10% coverage):
Number of contigs: 60
Largest contig: 14272 bp
N50: 2194 contigs
Total length: 103048 bp
SPAdes (careful):
Number of contigs: 34
Largest contig: 90035 bp
N50: 76572 contigs
Total length: 193763 bp
SPAdes (uncareful):
Number of contigs: 34
Largest contig: 90035 bp
N50: 76700 contigs
Total length: 193891 bp
SPAdes (10% coverage and careful):
Number of contigs: 8
Largest contig: 90035 bp
N50: 90035 contigs
Total length: 169704 bp
SPAdes (10% coverage and uncareful):
Number of contigs: 7
Largest contig: 90035 bp
N50: 90035 contigs
Total length: 169705 bp
We are unsure how to interpret which of these assemblies are the best based on these metrics, but the genome of the phage is expected to be 80-90 kb in total. None of the SPAdes assemblies fall within this range. Out of the Velvet assemblies only the 21 k-mer with 10% coverage fall with in the expected genome size. But the best assembly still remains to be discussed.